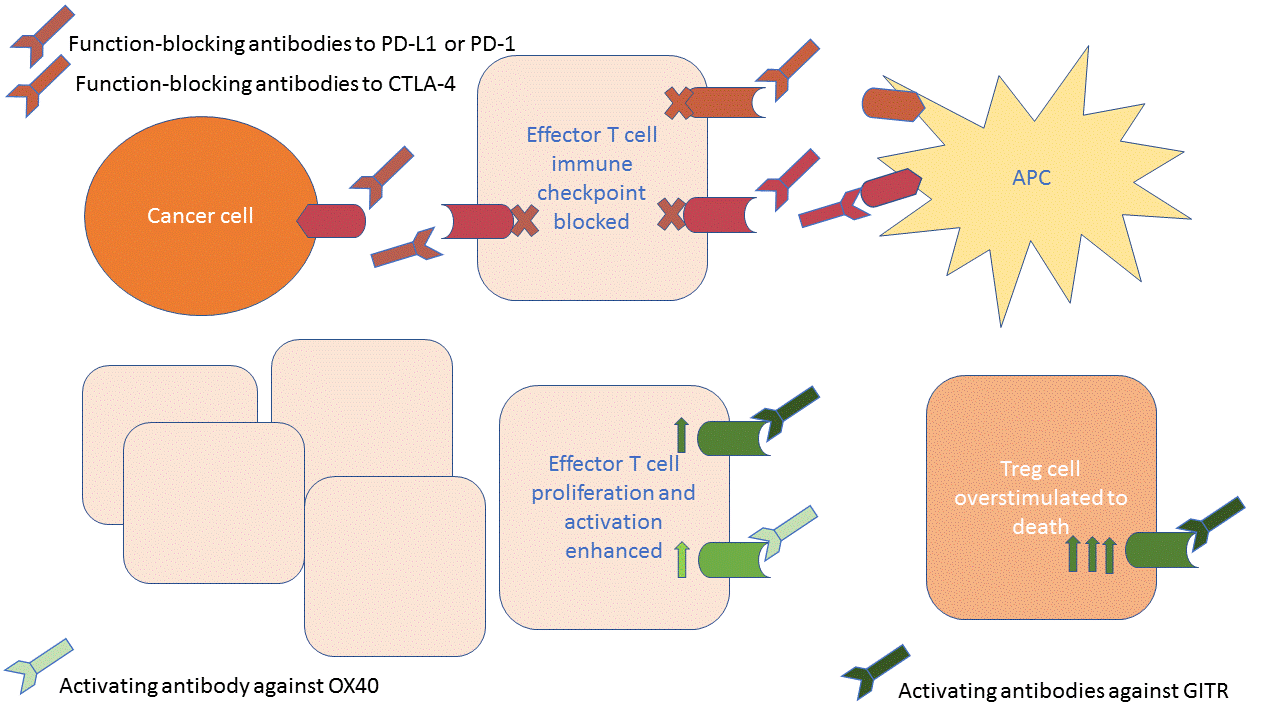

Some cancer immunotherapy drugs turn off a signaling pathway; others turn on a signaling pathway (1). FDA-approved immunotherapy drugs that target the PD-L1/PD-1 or CTLA-4/B-7 immune checkpoints function as antagonists to block immune signaling pathways. Investigational immunotherapy drugs that target GITRL/GITR function as agonists to activate an immune signaling pathway. Investigational drugs that target OX40L/OX40 also function as agonists to activate another immune signaling pathway.
Although each of the drugs is an antibody biologic agent, whether they activate or block the activity of their target is different, as is exactly which pair of the interacting partners can be targeted (Figure 1).
Although PD-L1 can be present on cancer cells or an antigen-presenting cells (APCs), PD-L1 and PD-1 interact selectively with each other. Thus, either protein can be targeted by function-blocking antibodies. For the CTLA-4/B-7 pathway, the B-7 proteins have other partners and those other interactions are beneficial in making the T cells attack the cancer. Thus, the function-blocking antibodies must target CTLA-4, not the B-7 proteins. This leaves the B-7 proteins on the APCs free to interact with stimulatory receptors on the T cells.
For the GITRL/GITR and OX40L/OX40 pathways, the goal is activation of these receptors. Therefore, the drugs are activating antibodies that target the receptors (GITR or OX40), not their matching ligands (GITRL or OX40L). In this case, the activating antibodies function as ligands, stimulating the receptor even if the natural ligand is not present.
Although the drugs targeting GITRL/GITR or OX40L/OX40 function as agonists, their anti-cancer mechanisms are quite different. OX40 functions on effector T cells to enhance their activity and proliferation. OX40 also functions on regulatory T cells (Tregs) to suppress their activity. Treg cells function as immunosuppressive cells that limit the activity of effector T cells. Clinical trials are in progress for OX40-targeted therapy.
GITR is also present on both effector T cells and Treg cells. GITR is a stimulatory receptor on both kinds of T cells. Experiments in mice indicated the importance of both the effector T cells and Tregs in mediating the anti-tumor response to GITR-activating antibodies (2, 3). These studies showed a decrease in the amount of activated Tregs, a reduction in markers of T cell exhaustion on the effector T cells, and an increase in activated effector T cells. Stimulation of Tregs with either GITRL or an activating antibody results in cell death, whereas the effector T cells do not show this toxic response. This difference in response may relate to the difference in the abundance of GITR on these two types of T cells: GITR is very abundant on Tregs and less abundant on effector T cells.
Clinical trials are underway for GITR-targeted therapies. A first-in-human trial with the GITR-activating antibody TRX-518 (4) showed a reduction in both activated GITR+ Tregs that appeared to result from both a decrease in their differentiation and an increase in their death. The results suggested that GITR+ activated Tregs may not only serve as a biomarker of GITR-activating antibody response but may also be a relevant target of these drugs.
The examples highlighted here show how the molecular targets and the function of the drugs needs to be carefully tuned to the output of the immune pathway. Sometimes both the ligand and the receptor can be useful targets as exemplified by antibodies that block either PD-L1 or PD-1. Sometimes only one of the interacting proteins is an appropriate target as in the CTLA-4, OX40, and GITR pathways. The drugs either need to block a signal or enhance a signal. Sometimes the consequence of the drug depends on the cell responding. For GITR-targeted therapies, Tregs are stimulated to death, whereas effector T cells become more active and functional. As always, the context is key to effective development of therapeutic strategies.
Related Reading
- W. C. M. Dempke, K. Fenchel, P. Uciechowski, S. P. Dale. Second- and third-generation drugs for immune-oncology treatment- The more the better? Eur. J. Cancer 74, 55-72 (2017). PubMed
- D. A. Schaer, S. Budhu, C. Liu, C. Bryson, N. Malandro, A. Cohen, H. Zhong, X. Yang, A. N. Houghton, T. Merghoub, J. D. Wolchok, GITR pathway activation abrogates tumor immune suppression through loss of regulatory T-cell lineage stability. Cancer Immunol. Res. 1, 320-331 (2013). PubMed
- A. E. Mahen, S. Mauze, B. Joyce-Shaikh, J. Xia, E. P. Bowman, A. M. Beebe, D. J. Cua, R. Jain, Dual roles for regulatory T-cell depletion and costimulatory signaling in agonistic GITR targeting for tumor immunotherapy. Cancer Res. 77, 1108-1118 (2017). PubMed
- R. Zappasodi, Y. Li, M. Abu-Akeel, J. Qi, P. Wong, C. Sirard, M. Postow, D. A. Schaer, W. Newman, H. Koon, V. Velcheti, M. K. Callahan, J. D. Wolchok, T. Merghoub, CT018: Intratumor and peripheral Treg modulation as a pharmcodynamic biomarker of the GITR agonist antibody TRX-518 in the first in-human trial. AACR Annual Meeting 2017 http://www.abstractsonline.com/pp8/#!/4292/presentation/12335
Also of Interest in BioSerendipity
Understanding Immune Checkpoint Pathways to Improve Patient Response
Immune Checkpoint Inhibitor Super Responders
Using Flu Vaccines to Treat Cancer
Intratumor Immunotherapy Kills Injected and Distant Tumors
Can you be vaccinated against yourself?
Cite as: N. R. Gough, Immunotherapy antagonists and agonists. BioSerendipity (25 May 2017). https://www.bioserendipity.com/immunotherapy-antagonists-and-agonists/