

Better understanding of the molecular mechanisms that control the adaptive immune system will lead to better immunotherapies for cancer patients or patients with other diseases related to inappropriate immune responses. Two of the processes that inhibit T cell function are called immune checkpoint pathways. New research reveals that these pathways involve many cells of the immune system, not just the T cells that are “turned off” by these pathways. With this new information, the right subset of patients that will respond well to drugs that prevent immune checkpoint signaling can be identified and new ways to expand the number of patients that will respond can be developed.
At least two immune checkpoint pathways function at the level of the effector CD8+ T cell (cytotoxic T cell), which is the type of adaptive immune cell that can kill a tumor cell, damaged cell, or infected cell. One pathway involves interactions between proteins on the T cell (PD-1) and on the target cell (PD-L1) and prevents the T cell from destroying the target cell. Cancer cells use PD-L1 to trigger this immune checkpoint response, tricking the T cell into behaving as if the cancer cell is a normal cell that should not be attacked. Oddly, the amount of PD-L1 or PD-1 that can be detected in tumor samples is not necessarily a good indicator of whether the patient will respond to immune checkpoint inhibitors.
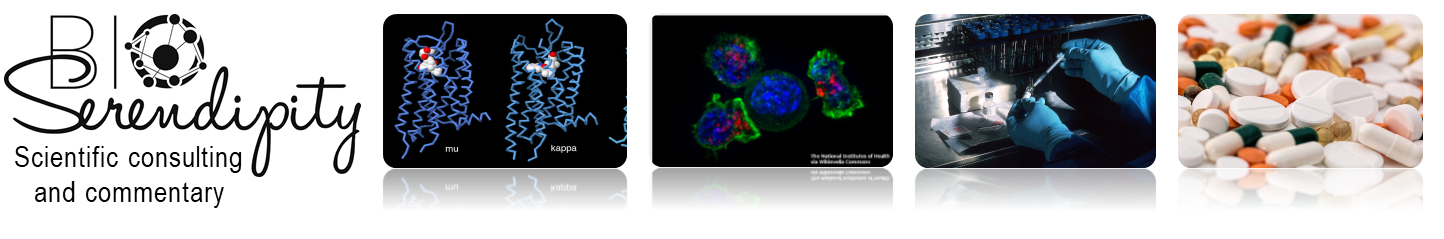
Other cells of the immune system change in response to signals produced by cancer cells or by the inflammation that is associated with cancer. Some of these changes activate a second immune checkpoint pathway, which is mediated by the protein CTLA-4 on the T cell. CTLA-4 is activated by proteins collectively referred to as B7 (also called CD80 and CD86) on another type of immune cell, antigen-presenting cells (APCs) (Figure 1). Compared with the PD-L1/PD-1 pathway, this second pathway is complex and context dependent.
Whereas PD-L1 is the only protein known to activate the PD-1 pathway; CTLA-4 is not the only T cell molecule that binds B7 proteins. B7 proteins can bind to CTLA-4 to turn off the T cell or B7 can bind to another T cell protein called CD28 to help activate T cells. In the context of cancer, the goals are to block the CTLA-4 interaction and promote the CD28 interaction. Thus, drugs aimed at blocking this second immune checkpoint inhibitor pathway must target CTLA-4 and not the B7 proteins. In contrast, drugs that block the PD-L1/PD-1 pathway can target either of PD-1 or PD-L1 (thus these are often referred to as PD-1/PD-L1 inhibitors). For either type of drug, the mechanism of action must be inhibition of the pathways mediated by CTLA-4 or PD-1.
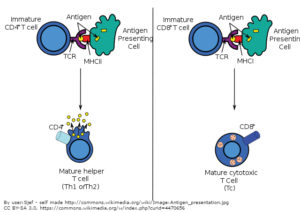
Biologic drugs that block these immune checkpoint pathways have the potential to reactivate the immune response to cancer (Figure 2). Keytruda (pembrolizumab) and Opdivo (nivolumab) are antibody drugs that bind and inhibit PD-1; Tecentriq (atezolizumab), Bavencio (aveluman), and Imfinzi (durvalumab) are antibody drugs that bind and inhibit PD-L1; and Yervoy (ipilimumab) is an antibody drug that binds and inhibits CTLA-4.
So far, none of the drugs reactivate only the cancer-targeted T cells, thus the drugs can result in the attack of normal cells by T cells (autoimmunity) or can exacerbate immune responses to infection or vaccination. Furthermore, not all patients exhibit a reduction in cancer when treated. Thus, it is critical to understand how to identify the patients most likely to respond so that fewer patients become exposed to the potential adverse effects caused by these drugs. It is also crucial to understand how immune checkpoint inhibitors change the immune response not only toward the cancer cells but the response induced by other medical interventions, such as vaccination, or by natural events, such as infection.
…none of the drugs reactivate only the cancer-targeted T cells…
T Cell State in the Response to PD-L1/PD-1 Inhibitors
One of the most perplexing aspects of trials involving the drugs that target the PD-L1/PD-1 pathway is that a good response does not necessarily correlate with the amount of PD-L1 on the tumor cells based on biopsy results. Philip and colleagues provide evidence that the responsiveness to PD-L1/PD-1 inhibitors depends on the state of the T cells themselves, not just whether the CD8+ T cells had a lot of PD-1 (PD-1high) and the tumor had a lot of PD-L1. In a mouse cancer model, they found that even the tumor-specific CD8+ T cells that were PD-1high exhibited two distinct chromatin states (Figure 3). The first state produced a dysfunctional T cell that could be reactivated; whereas T cells of the second state could not be reactivated. With current approaches, state 1 was a reversible dysfunctional condition and state 2 was an irreversible dysfunctional condition. This suggests that it is not enough to analyze the tumor sample for the number of CD8+ T cells and the amount of PD-1 on those cells.
However, measuring epigenetic status is technically challenging and not readily implemented for patient care. Thus, the authors sought surface protein markers that could be used to determine if a patient’s PD-1high tumor-associated T cells were state 1 or state 2 and thus were likely to respond to checkpoint inhibitors and recognize the cancer cells. The authors identified additional proteins on mouse tumor-specific CD8+ T cells that differentiated the reversible state 1 cells from the irreversible state 2 cells. Reversibly dysfunctionally programmed T cells were PD-1high, CD38low, CD101low and the irreversibly dysfunctional T cells were PD-1high, CD38high, CD101high, and CD5low. Melanoma and lung cancer patients had a population of PD-1high CD8+ T cells that fit the marker profile for state 2 (CD38high, CD101high, and CD5low), suggesting that some of their tumor-infiltrating lymphocytes were irreversibly dysfunctional. Thus, future studies should assess if administration of drugs that alter epigenetic reprogramming along with or prior to the PD-L1/PD-1 checkpoint inhibitor may be beneficial and whether patients with the reversibly reprogrammed T cells are better candidates for immune checkpoint inhibitor therapy than those with mostly state 2 T cells.
APCs in the Response to PD-L1/PD-1 Inhibitors
Not only are the CD8+ T cells and cancer cells important for the anti-tumor response, but other cells also play a role. Sometimes the response correlates with the amount of PD-L1 on the tumor-infiltrating immune cells, rather than on the tumor cells. A pair of studies in Science sheds light on additional molecular requirements for effective responses to PD-1/PD-L1 inhibitors and suggests that APCs may be a population of tumor-infiltrating cells that dictate responsiveness to these inhibitors.
Kamphorst and colleagues examined a mouse model cancer. They measured the amount of proliferating CD8+ T cells that recognized the cancer to determine the response to the PD-1 or PD-L1 inhibitor. Unexpectedly, the increase in these proliferating CD8+ T cells caused by the inhibitor depended on the T cell protein CD28 and the protein B7, which delivers a costimulatory signal to T cells. In the mouse cancer model, the antibody against PD-L1 reduced tumor size and increased survival, but blocking the CD28-B7 interaction prevented this anticancer response.
…inhibitor [response] depended on the T cell protein CD28 and the protein B7…
The relevance of the mouse findings was tested by analyzing circulating CD8+ T cells in non-small cell lung cancer patients who had received PD-1-targeted immune checkpoint inhibitor. In the patients that had an increase in the number of proliferative and activated CD8+ T cells after treatment, these CD8+ T cells were also positive for CD28. Thus, just like in the mouse cancer model, the ability to respond to PD-1 immune checkpoint therapy correlated with and may depend on the number of CD8+ T cells that can be costimulated by CD28. Because the molecule that binds and activates CD28 is present on APCs, not tumor cells, this study also indicates that these other immune cells contribute to the T cell response to PD-1 inhibitors.
In a biochemical study, Hui and colleagues provide a molecular explanation for the dependency of PD-L1/PD-1 inhibition on CD28. PD-1 specifically interacted with the protein phosphatase SHP2. The co-receptor CD28, instead of the T cell receptor, was the protein most efficiently dephosphorylated in the reconstituted system. Experiments with T cells confirmed that CD28 was the preferred target that became dephosphorylated in response to PD-1 activation. Thus, by targeting CD28, PD-1 signaling shuts down T cells. These two studies suggest that PD-L1 on tumor-infiltrating APCs may block costimulatory signals presented by these cells to the T cells.
Thus, by targeting CD28, PD-1 signaling shuts down T cells.
Tumor-associated macrophages in the response to PD-L1/PD-1 inhibitors
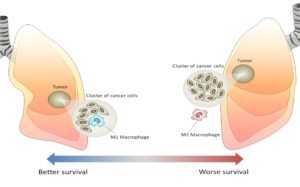
A study by Gordon and colleagues brings tumor-associated macrophages (TAMs) into the mechanism of action of PD-L1/PD-1 inhibitors. Macrophages are white blood cells that produce signals that stimulate or inhibit inflammation. They also function as APCs for T cells. Depending on the signals that these cells release, the proteins on their surface, and the presence of other proteins that serve as markers, macrophages are classified as M1 or M2. M1 macrophages are considered inflammatory and release proinflammatory signals that activate and recruit other immune cells. M2 macrophages are considered anti-inflammatory and promote healing of damaged or injured tissue. In the context of cancer, M1 macrophages are considered anti-tumor and M2 macrophages are considered pro-tumor (Figure 4).
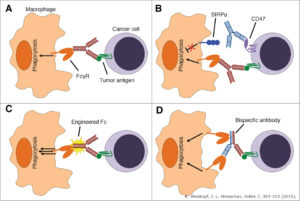
Another key function of macrophages is engulfing cellular debris. As such, these cells serve as the “clean up” crew by “eating” (phagocytosing) pathogens, infected cells, damaged cells, and cancer cells. Both M1 and M2 macrophages can be highly phagocytic. Indeed, antibody-based cancer therapies may promote phagocytosis of the cancer cells with the proteins recognized by the antibodies (Figure 5). However, the study by Gordon and colleagues showed that the presence of PD-1 on tumor-associated macrophages (TAMs) inhibits their ability to phagocytose tumor cells.
The authors used mouse cancer models to show that the amount of PD-1 present on TAMs increased over time and with tumor size. These PD-1-positive TAMs were of the M2 type and did not phagocytose tumor cells effectively. The authors used engineered mouse cancer models to explore the role of PD-1 on the TAMs and PD-L1 on the cancer cells in tumor growth. These mouse studies showed that the tumor growth and impaired phagocytosis of tumor cells by the PD-1-positive macrophages required the cancer cells to have PD-L1. Furthermore, giving these mice treatments that blocked the PD-L1/PD-1 interaction reduced tumor growth in a way that depended on the TAMs.
Like other immune cells, macrophages become “activated” and the body has mechanisms for preventing excessive activity. The work of Gordon and colleagues reveals the PD-L1/PD-1 pathway as one of these. However, another macrophage checkpoint pathway is already known. This one involves the protein SIRPα (signal-regulatory protein α) on the macrophage and CD47 on the target cell. CD47 tells the macrophage that the cell is “self” and should not be eaten. Tumor cells can increase the amount of CD47 and thus prevent phagocytosis by macrophages. Indeed, antibody-based therapies targeting CD47 or SIRPα are in clinical trials for cancer. Gordon and colleagues combined a CD47-targeted antibody with a peptide that blocks PD-L1 function in the mouse cancer model and found that combined therapy reduced tumor size and prolonged survival more than either therapy alone.
These results may explain why assessing the abundance of PD-L1 on the cancer cells is inadequate to predicting patient responsiveness. A detailed picture of the status of the tumor-associated immune system (Figure 6) needs to be determined too.
Key Points for the Future of PD-L1/PD-1 Immune Checkpoint Inhibitors
- Activation of the PD-L1/PD-1 checkpoint in T cells may come from PD-L1 on tumor cells or on other immune cells.
- The presence of PD-1 on the T cells needs to be assessed along with markers of the dysfunctional state to the cells to identify the best responders.
- The PD-L1/PD-1 immune checkpoint pathway functions in both T cells and macrophages.
- Drugs that inhibit the PD-L1/PD-1 may reactivate T cells to kill the cancer cells and macrophages to clear the cancer cells by phagocytosis.
- Combining drugs that influence epigenetic remodeling with PD-L1/PD-1 inhibitors may be beneficial.
- Combining drugs that impair other macrophage immune checkpoints with PD-L1/PD-1 inhibitors may be beneficial.
Read the next commentary article in this series:
Related Reading
ELCC 2017: Patients with lung cancer treated with PD-1/PD-L1 checkpoint inhibitors may experience adverse events after influenza vaccination. The ASCO Post (27 April 2017).
Immune checkpoint inhibitors to treat cancer. American Cancer Society (updated 3 February 2017). https://www.cancer.org/treatment/treatments-and-side-effects/treatment-types/immunotherapy/immune-checkpoint-inhibitors.html
I. Buchbinder, A. Desai, CTLA-4 and PD-1 pathways: Similarities, differences, and implications of their inhibition. Am. J. Clin. Oncology 39, 98–106 (2016). DOI: 10.1097/COC.0000000000000239 PubMed
E. Conway, The many faces of macrophages in cancer. Atlas of Science (17 February 2016) https://atlasofscience.org/the-many-faces-of-macrophages-in-lung-cancer/
S. R. Gordon, R. L. Maute, B. W. Dulken. G. Hutter, B. M. George, M. N. McCracken, R. Gupta, J. M. Tsai, R. Sinha, D. Borey, A. M. Ring, A. J. Connolly, R. L. Weissman, PD-1 expression by tumour-associated macrophages inhibitors phagocytosis and tumour immunity. Nature (17 May 2017). DOI: 10.1038.nature22396 PubMed
E. Hui, J. Cheung, J. Zhu, X. Su, M. J. taylor, H. A. Wallweber, D. K. Sasmal, J. Huang, J. M. Kim, I. Mellman, R. D. Vale, T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science (9 March 2017). DOI: 10.1126/science.aaf1292 PubMed
O. Kamphorst, A. Wieland, T. Nasti, S. Yang, R. Zhang, D. L. Barber, B. T. Konieczny, C. Z. Daugherty, L. Koenig, K. Yu, G. L. Sica, A. H. Sharpe, G. J. Freeman, B. R. Blazar, L. A. Turka, T. K. Owonikoko, R. Pillai, S. S. Ramalingam, K. Araki, R. Ahmed, Rescue of exhausted CD8 T cells by PD-1–targeted therapies is CD28-dependent. Science (9 March 2017). DOI: 10.1126/science.aaf0683 PubMed
M. Philip, L. Fairchild, L. Sun, E. L. Horste, S. Camara, M. Shakiba, A. C. Scott, A. Viale, P. Lauer, T. Merghoub, M. D. Hellmann, J. D. Wolchok. C. S. Leslie, A. Schietinger, Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature (17 May 2017). DOI: 10.1038/nature22367. PubMed
K. Weiskopf, I. L. Weissman, Macrophages are critical effectors of antibody therapies for cancer. mAbs 7, 303-310 (2015). PubMed
Cite as: N. R. Gough, Understanding immune checkpoint pathways to improve patient response. BioSerendipity (24 May 2017). https://www.bioserendipity.com/2017/05/24/understanding-im…patient-response/